New features of AbiPy v0.7¶
M. Giantomassi and the AbiPy group¶
9th international ABINIT developer workshop
20-22nd May 2019 - Louvain-la-Neuve, Belgium

- These slides have been generated using jupyter, nbconvert and revealjs
- The notebook can be downloaded from this github repo
- To install and configure the software, follow these installation instructions
Use the Space key to navigate through all slides.
![]()
What is AbiPy?¶
Python package for:¶
- Generating ABINIT input files automatically
- Post-processing output results (netcdf and text files)
- Interfacing ABINIT with external tools (e.g. Vesta)
- Creating and executing workflows (band structures, DFPT, $GW$, BSE…)
Why python?¶
- Easy to use and to learn
- Great support for science (numpy, scipy, pandas, matplotlib …)
- Interactive environments (ipython, jupyter notebooks, GUIs)
- More powerful and flexible than Fortran for implementing the high-level logic needed in modern ab-initio workflows
- pymatgen ecosystem and the materials project database…
How to install AbiPy¶
Using pip and python wheels:
pip install abipy --user
Using conda (recommended):
conda install abipy --channel abinit
From the github repository (develop mode):
git clone https://github.com/abinit/abipy.git
cd abipy
python setup.py develop
For further info see http://abinit.github.io/abipy/installation.html
AbiPy documentation with galleries of matplotlib examples and workflows¶
In [57]:
%embed https://abinit.github.io/abipy/index.html
Out[57]:
Jupyter notebooks with examples and lessons inspired by the official tutorials¶
In [58]:
%embed https://nbviewer.jupyter.org/github/abinit/abitutorials/blob/master/abitutorials/index.ipynb
Out[58]:

Since AbiPy is not restricted to high-throughput, we'll show how to use the terminal to analyze calculations
Well, a python script would be much more flexible but the goal here is to show that one can replace grep, vim, gnuplot with AbiPy
No perl scripts were harmed in the making of this notebook
Command line interface¶
- abiopen.py ➝ Open output files inside ipython or print/visualize file
- abistruct.py ➝ Operate on crystalline structures read from file
- abicomp.py ➝ Compare multiple files (i.e. convergence studies)
- abiview.py ➝ Quick visualization of output files
- abinp.py ➝ Generate input files for typical calculations
Documentation¶
abistruct.py --helpfor manpageabistruct.py COMMAND --helpfor help aboutCOMMAND
HTML documentation available at http://abinit.github.io/abipy/scripts/index.html
Examples¶
abistruct.py spglib si_scf_GSR.nc
abistruct.py convert si_scf_GSR.nc -f cif
abiopen.py si_scf_GSR.nc --print
and many more...¶
Print results stored in FILE (more than 45 file extensions supported)¶
In [59]:
!abiopen.py si_scf_GSR.nc --print
To produce a predefined set of matplotlib figures, use:¶
abiopen.py mgb2_kpath_FATBANDS.nc --expose --seaborn
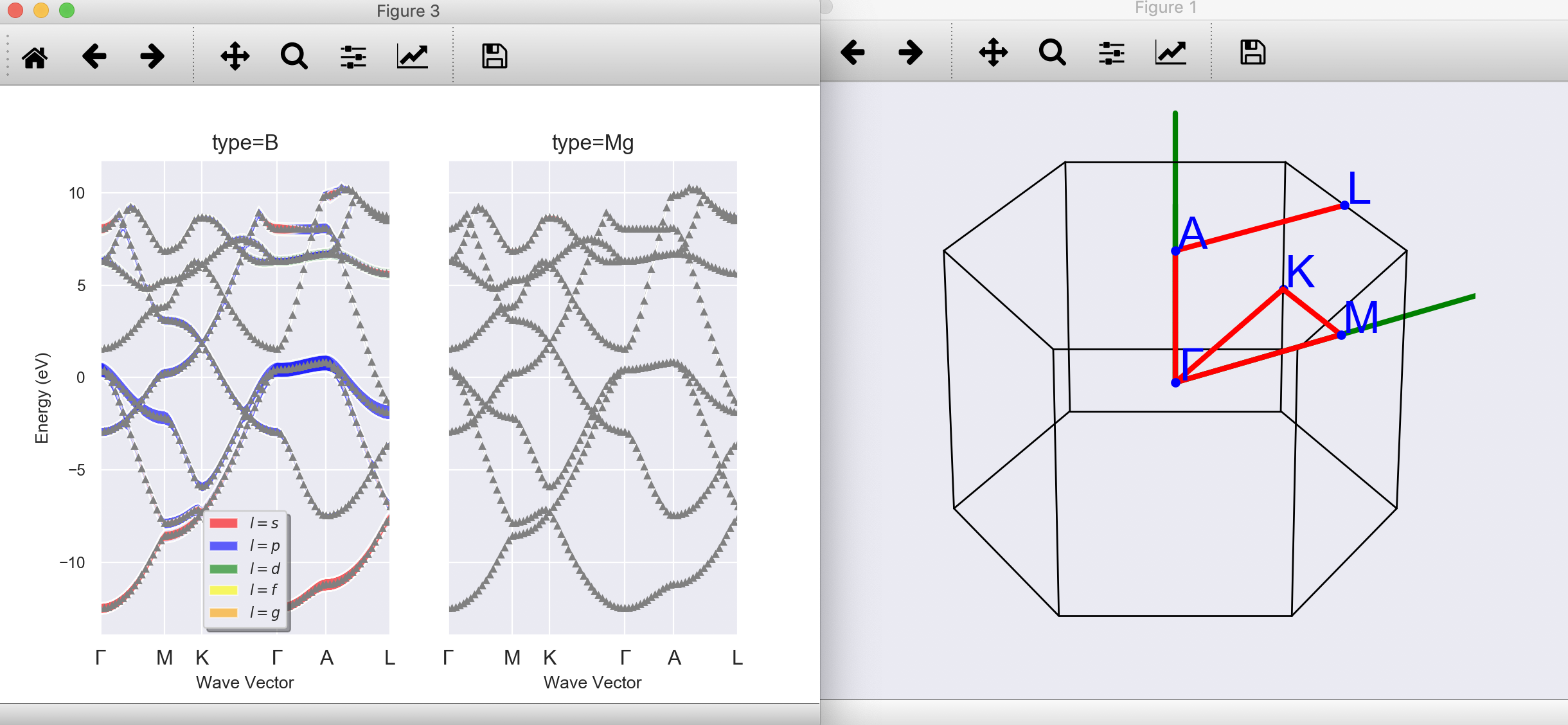
Replace --expose with --notebook to generate a jupyter notebook with predefined python code¶
abiopen.py supports output files (note abo extension):¶
In [60]:
!abiopen.py gs_dfpt.abo -p
and log files as well:¶
In [61]:
!abiopen.py run.log -p
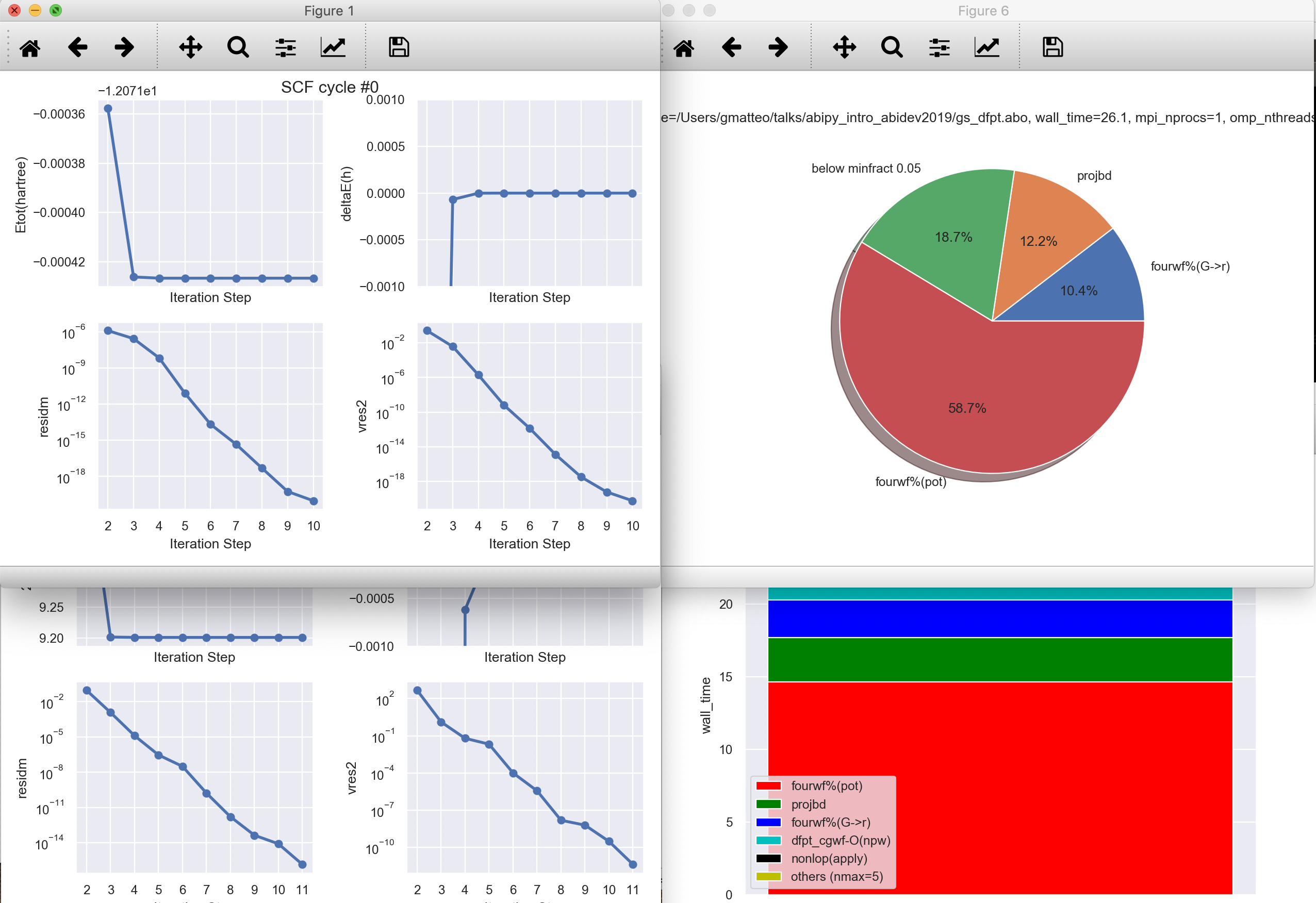

It works with any file providing a structure object (.nc, .abi, .cif …)¶
Convert structure from netcdf format to CIF (abivars, xsf, poscar, qe, siesta, wannier90, json)¶
In [62]:
!abistruct.py convert si_scf_GSR.nc -f cif
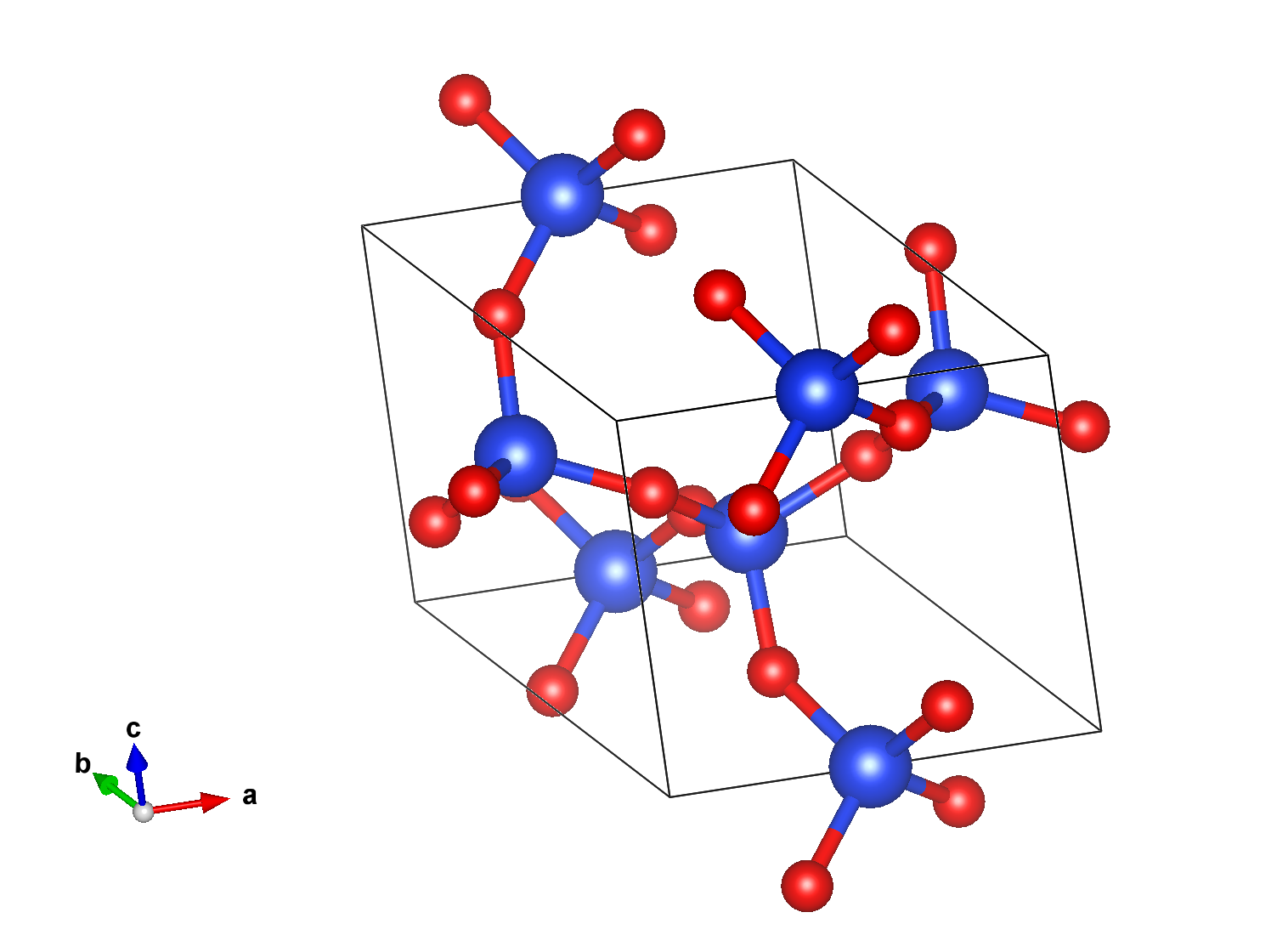
Connect to the materials project database to find similar structures.¶
In [66]:
!abistruct.py mp_match si_scf_GSR.nc
To search by chemical system or formula (no FILE required)¶
In [67]:
!abistruct.py mp_search LiF
Need to call anaddb to compute and visualize ph-bands and DOS from DDB?¶
abiview.py ddb ZnSe_hex_qpt_DDB --seaborn
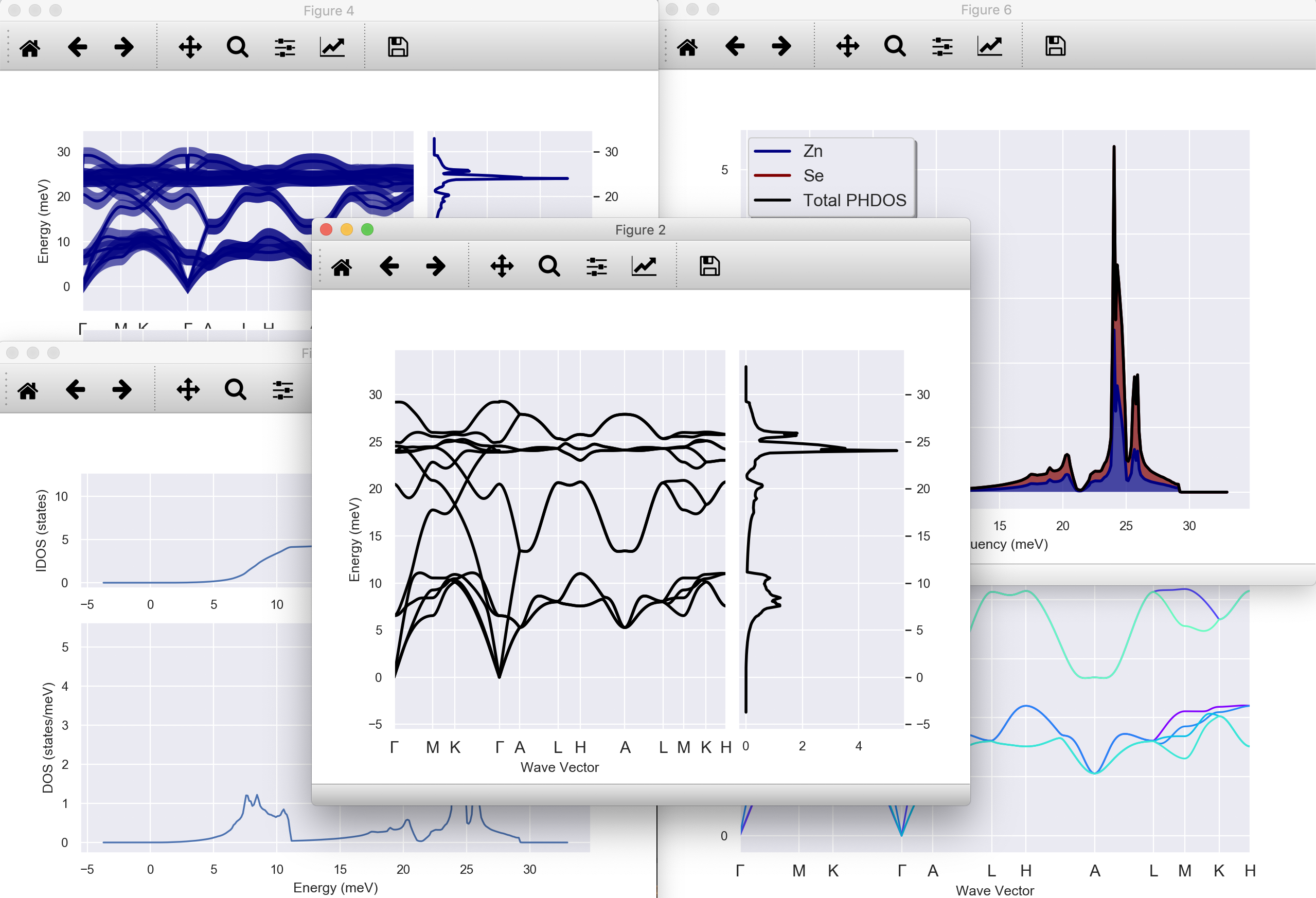
Add --phononwebsite to visualize data on the phononwebsite by Henrique¶

To compare multiple structures:¶
In [68]:
!abicomp.py structure *.cif si_nscf_GSR.nc `find . -name *_DDB`
Note shell wildcard characters and Unix find inside backticks (bash rocks!)¶
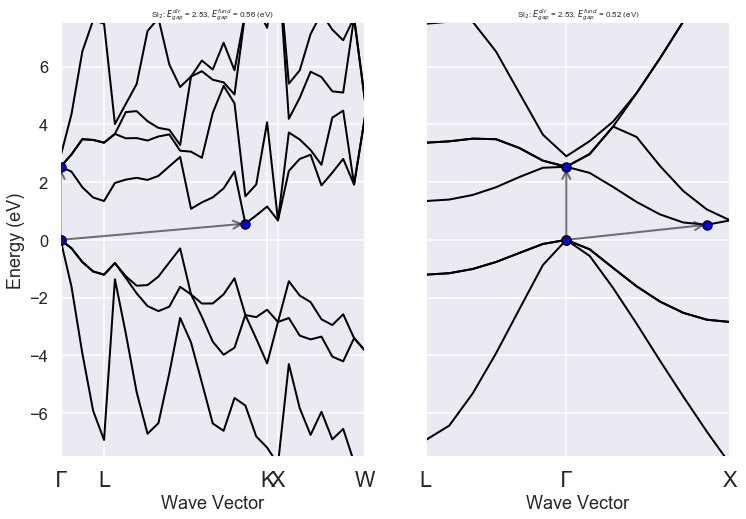
In [70]:
paths = [
"mgb2_888k_0.01tsmear_DDB",
"mgb2_888k_0.04tsmear_DDB",
"mgb2_121212k_0.01tsmear_DDB",
"mgb2_121212k_0.04tsmear_DDB",
]
paths = [os.path.join(abidata.dirpath, "refs", "mgb2_phonons_nkpt_tsmear", f)
for f in paths]
robot = abilab.DdbRobot()
for i, path in enumerate(paths):
robot.add_file(path, path)
In [71]:
# Define function to change labels:
func = lambda ddb: "nkpt: %s, tsmear: %.2f" % (
ddb.header["nkpt"], ddb.header["tsmear"])
robot.remap_labels(func)
robot
Out[71]:
Now we can build a dataframe with the most important parameters:¶
In [72]:
robot.get_params_dataframe()
Out[72]:
and check that all DDBs have been computed with the same crystalline structure:¶
In [73]:
robot.get_lattice_dataframe()
Out[73]:
To analyze the effect of k-sampling/smearing on the vibrational properties:¶
In [74]:
# Invoke anaddb and store results
r = robot.anaget_phonon_plotters(nqsmall=2)
r.phbands_plotter.gridplot_with_hue("tsmear", with_dos=True);


Input file for band structure calculation + DOS¶
- GS run to get the density
- NSCF run along high-symmetry k-path
- NSCF run with k-mesh to compute the DOS
In [77]:
multi = abilab.ebands_input(structure="si.cif",
pseudos="14si.pspnc",
ecut=8,
spin_mode="unpolarized",
smearing=None,
dos_kppa=5000)
multi.get_vars_dataframe("kptopt", "iscf", "ngkpt")
Out[77]:
To build an input for SCF+NSCF run with (relaxed) structure from the materials project database:¶
abinp.py ebands mp-149 abinp.py is handy for generating input files quickly but it cannot compete with the flexibility of the python interface:¶
In [79]:
def make_scf_input(ecut=2, ngkpt=(4, 4, 4)):
"""
Generate an `AbinitInput` to perform GS calculation for AlAs.
Args:
ecut: Cutoff energy in Ha.
ngkpt: k-mesh divisions
Return:
`AbinitInput` object
"""
gs_inp = abilab.AbinitInput(structure="AlAs.cif",
pseudos=["13al.pspnc", "33as.pspnc"])
# Set the value of the Abinit variables needed for GS runs.
gs_inp.set_vars(
nband=4,
ecut=ecut,
ngkpt=ngkpt,
nshiftk=4,
shiftk=[0.0, 0.0, 0.5, # This gives the usual fcc Monkhorst-Pack grid
0.0, 0.5, 0.0,
0.5, 0.0, 0.0,
0.5, 0.5, 0.5],
tolvrs=1.0e-10,
)
return gs_inp
Inside the notebook, one gets the HTML representation with links to the documentation:¶
In [80]:
make_scf_input()
Out[80]:
Moreover, from an AbiniInput one can easily build more complicate workflows:¶
In [81]:
def build_flow_alas_phonons():
"""
Build and return a Flow to compute the dynamical matrix on a (2, 2, 2) qmesh
as well as DDK and Born effective charges.
The final DDB with all perturbations will be merged automatically and placed
in the Flow `outdir` directory.
"""
from abipy import flowtk
scf_input = make_scf_input(ecut=6, ngkpt=(4, 4, 4))
return flowtk.PhononFlow.from_scf_input("flow_alas_phonons", scf_input,
ph_ngqpt=(2, 2, 2), with_becs=True)
Abipy will call Abinit to get the list of DFPT perturbations and…¶
In [82]:
flow_phbands = build_flow_alas_phonons()
flow_phbands.get_graphviz()
Out[82]:
Future developments¶
Post-processing tools¶
- Support for more netcdf files
- More post-processing tools for MD calculations
- More converters and interfaces for third-party applications
- Integrate AbiPy with jupyterlab to create a flexible graphical enviroment for Abinit exposing (part) of the python API
- Explore web-based technologies for data analysis and visualization (plotly, dash)
- Develop toolkit to build web apps powered by AbiPy and pymatgen to disseminate scientific results.
Continuous Integration¶
Use AbiPy programmatic interface to implement:
- Validation of parallel algorithms for np in range(1, N)
- Stress testing
- Benchmarks